Main Window
Vipster has a modular graphical interface.
Upon launch it shows one visualisation area and the Molecule widget to the left.
Details for these and other available widgets are listed below.
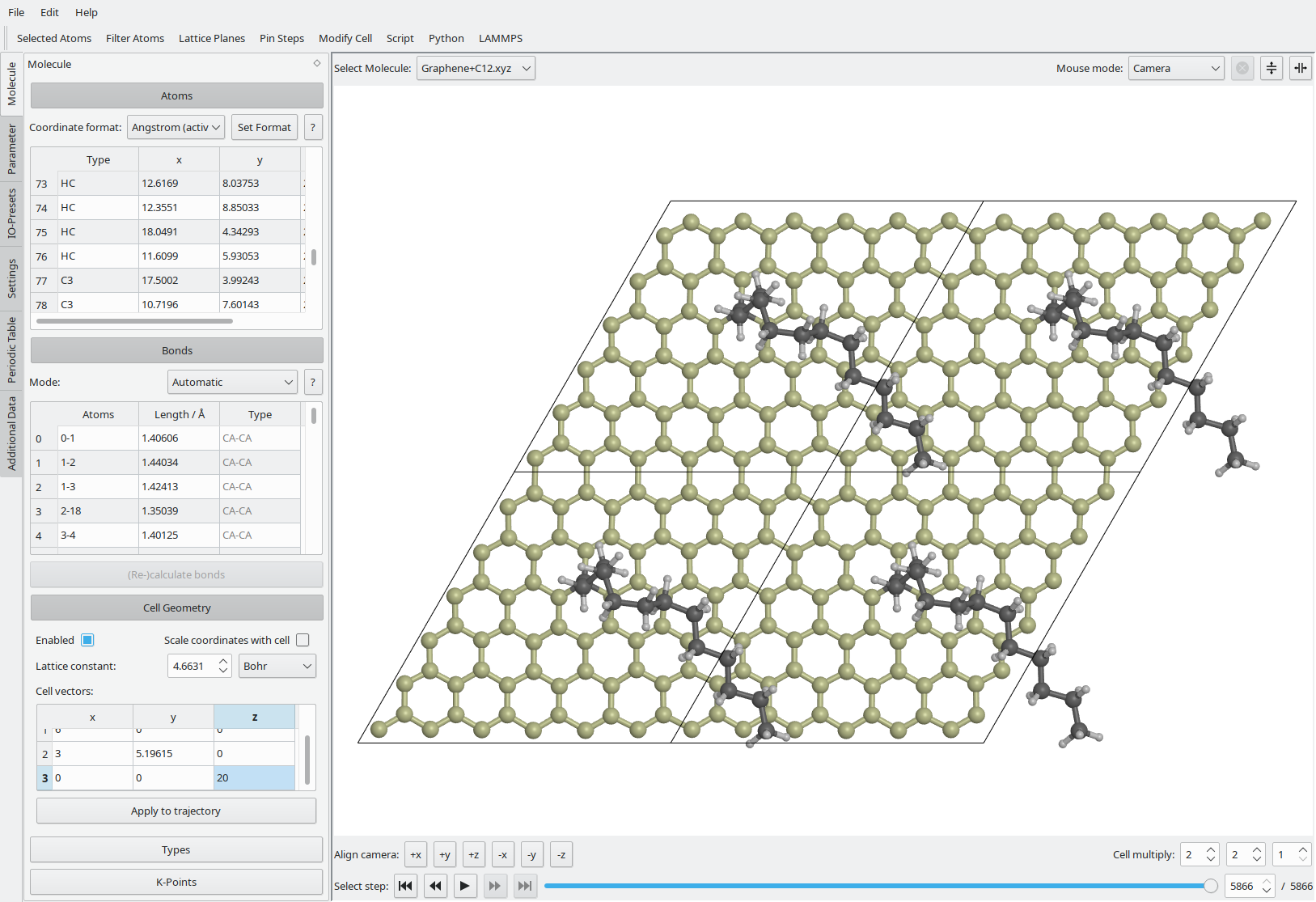
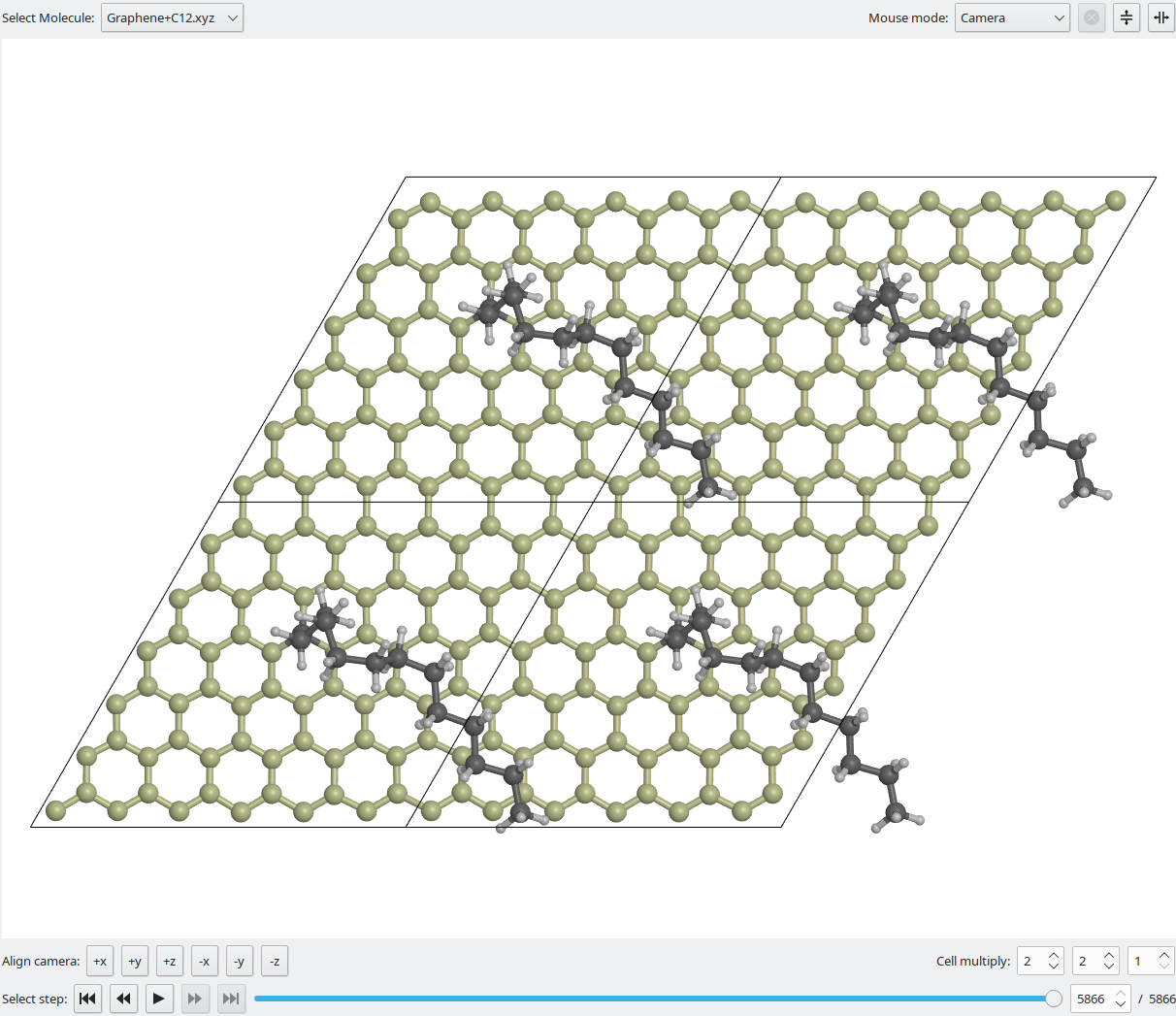
The main interaction area are the visualisation widgets.
Upon launch, one widget is displayed. Via the buttons in the top right corner, additional visualisation widgets can be added and removed as needed. Other widgets always act on the data selected in the active (highlighted) visualisation widget.
The Select Molecule dropdown menu shows the molecules read from files or created within Vipster. If a molecule contains multiple steps (e.g. a molecular dynamics trajectory), they can be selected via the button and slider controls at the bottom.
Multiple modes for mouse interaction can be selected:
- Camera will rotate, zoom and pan the camera.
- Select Atoms will enable to select single or multiple atoms by highlighting them.
- Modify Atoms will rotate or shift the molecule or currently selected atoms (if any).
- Modify Bonds (only active if bond mode is manual) will allow to create and remove bonds by dragging between two atoms.
If a molecule has a unit cell, the Cell multiply selectors can be used to display multiple repetitions of the cell.
Align camera will align the camera along the respective coordinate axis.
Main Widgets
On the left hand border, the main widgets can be selected. These widgets expose basic interaction with loaded data.
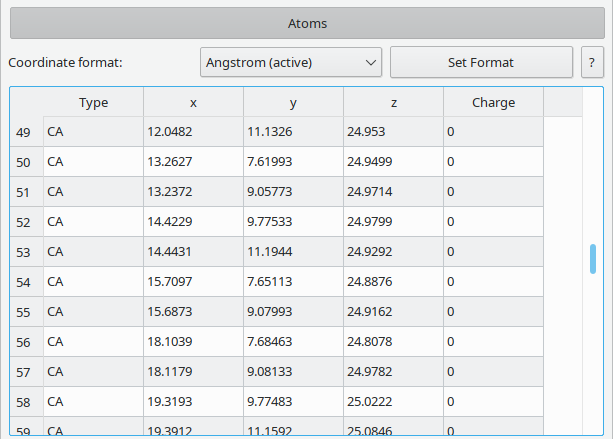
The properties of individual atoms can be modified via this table. By default it displays the type and position, other properties can be toggled by right clicking the table header.
Atom properties:
- Type given as an arbitrary user-defined string. Vipster tries to guess the (derived) atom type as described in the Type section.
- Coordinates, displayed in the chosen coordinate format.
- Forces, arbitrary format.
- Charge, arbitrary format.
- Visibility, toggle display of this atom.
- Constraints, toggle keeping certain parts of this atom's coordinates fixed.
The coordinates can be shown in multiple formats:
- Crystal: relative to cell dimension and cell vectors.
- Alat: relative to cell dimension.
- Ångström: absolute position given in 10-10m.
- Bohr: absolute position given in multiples of the Bohr radius (approx. 0.529Å).
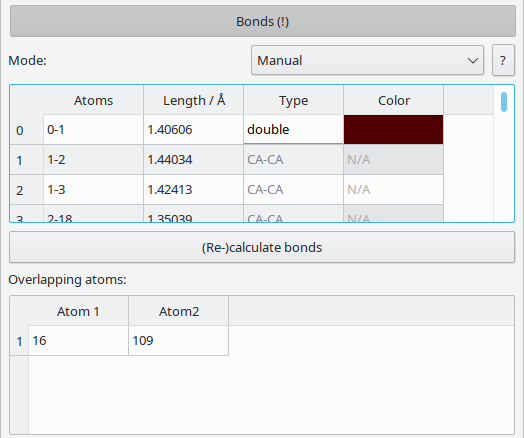
Bonds in Vipster are a connection between two atoms. There is no intrinsic notion of e.g. double bonds.
The bond mode can be set to either automatic oder manual.
In automatic mode, bonds are evaluated whenever the current structure changes. A bond will be created if the distance between two atoms is shorter than the sum of their type's bond cutoff radius times 1.1. The type of an automatically generated bond in turn is defined via the bonded atoms types.
Manual mode disables the automatic evaluation. If a parsed file contains pre-defined bonds, the molecule will default to manual bond mode. The user can apply the automatic evaluation mechanism above by pressing the (Re-)calculate bonds button. Bonds can also be added or removed on a per-bond basis via graphical selection. The automatically generated bond type can be replaced by supplying a user-defined string.
If the header of this section contains an exclamation mark (i.e. Bonds (!)), two or more atoms have to be found to overlap. Keep in mind that this may also refer to overlapping atoms under periodic boundary conditions.
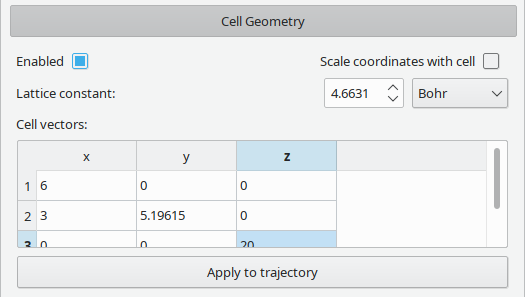
Vipster has intrinsic support for periodic structures.
A unit cell is defined as three unit-less vectors, scaled by a lattice constant having a unit.
If the Scale coordinates with cell box is checked, all changes to the unit cell will be applied to the contained atoms: If the lattice constant is doubled, an atom with an x-coordinate of 2Å will now sit at 4Å. The relative positioning inside the cell will be kept constant, however.
In a trajectory, each step can have its own unit cell. Via the Apply to trajectory button however, the current settings can be applied across the complete trajectory. This also respects the scaling setting.
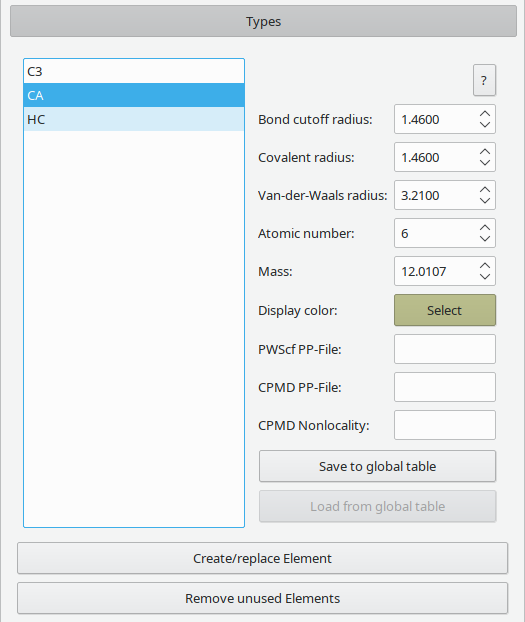
All atoms loaded into Vipster have a certain type, specifying multiple properties.
The type is changed by assigning an arbitrary name to an atom, either when reading from a file or by user interaction.
Types are stored in the molecule's periodic table. This table only contains types (previously) encountered in this molecule. If a type is not known, it will be looked up from the global periodic table by fuzzy matching against known atom types. During matching, trailing characters will be removed until a known type is encountered. If a name is purely numeric, it will be matched against the proton number. Types are shared across a trajectory, if applicable.
Via the widgets buttons, new types can be created and interchanged between the molecule's table and the global table. Types stored in the global table will be remembered by Vipster across multiple launches.
Description of the properties
- Bond cutoff radius: cutoff for automatic determination of bonds (defaults to covalent radius).
- Covalent radius: used for display (default).
- Van-der-Waals radius: alternatively used for display.
- Atomic number: used for type matching and in some file formats.
- Mass: used for type matching and in some file formats.
- Display color: used for representation, may contain transparency.
- PWScf PP-File: pseudopotential file to be used when writing PWScf input. See format for details.
- CPMD PP-File/Nonlocality: pseudopotential file and settings to be used when writing CPMD input. See format for details.
The Periodic table widget works exactly like this section of the Molecule widget, but controls the global periodic table.
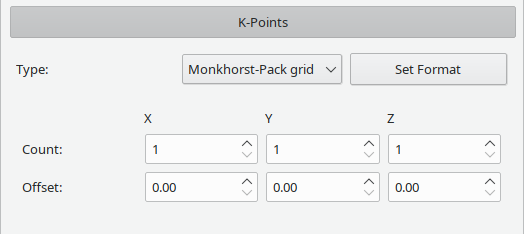
Quantum mechanical calculations of periodic systems often use a k-Point mesh to achieve good representations of periodic systems even with small unit cells.
Supported settings are:
- Gamma: only the k-Point at the origin of the brillouin zone will be used. Some codes have optimizations for this case.
- Monkhorst-Pack grid: will let the code generate an evenly spaced grid with count points along the corresponding vector of the brillouin zone. May be offset by certain vectors, see the documentation of your code for details.
- Discrete list of points: manually specify a number of k-Points to use. Crystal will tell your code to interpret the coordinates relative to the brillouin zone. Band will tell your code to generate a path along certain k-Points for band structure generation. See the documentation of your code for details and specific syntax.
Note that only the active format will be used when writing to a file.
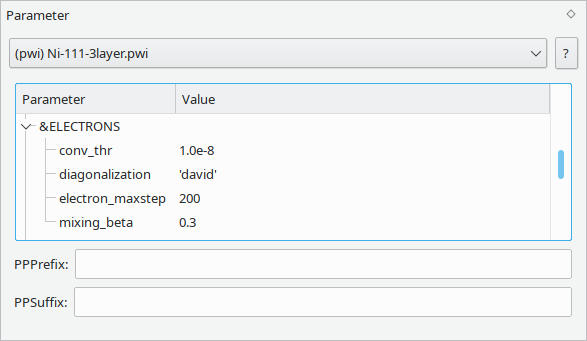
Simulation input consists of both the atomistic structure and parameters controlling various aspects of a calculation. Vipster encapsulates these Parameters and allows them to be reused in a mix and match fashion for easy setup of calculations.
Only files intended for input for a simulation software use and require parameter sets. Please refer to the parameters section of a specific format for details on the type and meaning of available settings.
When a suitable input file is read by Vipster, the corresponding parameter set will be extracted and loaded in this widget. Alternatively, an empty or previously saved parameter set can be loaded from the file menu.
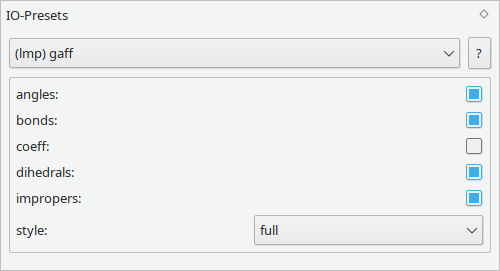
Similar to the parameters, IO Presets are used to control the generated files. They control the way in which data is formatted and written to a file, e.g. the format used to specify coordinates.
Please refer to the output preset section of a specific format for details on the type and meaning of available settings.
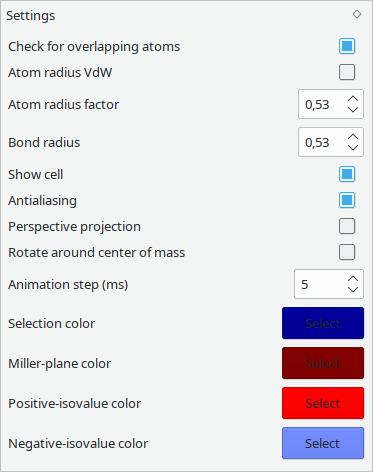
General behaviour and visualisation settings. Will be saved on exit.
- Check for overlapping atoms: notifies about overlapping atoms in the bonds section and on save. May cause slowdowns with large systems.
- Atom radius VdW: render atoms based on their van der Waals radius instead of the covalent radius.
- Atom radius factor: scales the radius before rendering.
- Bond radius: display radius for bonds.
- Show cell: toggle rendering of unit cell outlines.
- Antialiasing: toggle multisampling, may cause slowdowns on older GPUs or with large systems.
- Perspective projection: render with perspective instead of orthogonal projection.
- Rotate around center of mass: toggle between rotating around center of atoms instead of center of unit-cell.
- Animation step (ms): timestep of automatic stepping through a trajectory (with play button).
- Selection color: color of shell around selected atoms.
- Miller-plane color: color of plane set via Lattice Planes widget.
- Positive-/Negative-isovalue color: colors of isosurface generated via Additional data widget.
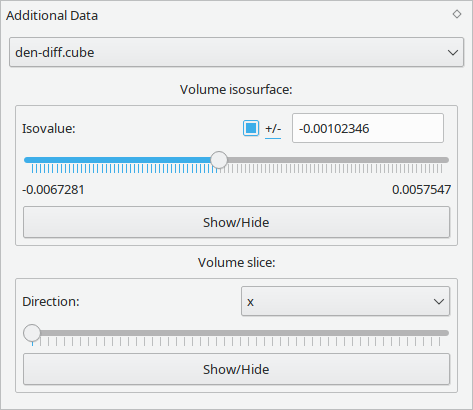
Data that is neither tied to the atomic structure nor the simulation setup is loaded as Additional Data. Currently, this refers to two and three-dimensional grids of scalar data. A rainbow-style color map can be used to display planar data.
Two-dimensional data grids can be loaded from XSF files and be displayed as a color mapped plane.
Three-dimensional data grids can be loaded from XSF and Cube files. They can be visualised either as a color mapped slice across the grid volume or as a isosurface at a certain isovalue.
Tool widgets
Optional tool widgets offer advanced interaction and modification options for molecular data.
Show situation dependent information about selected atoms.
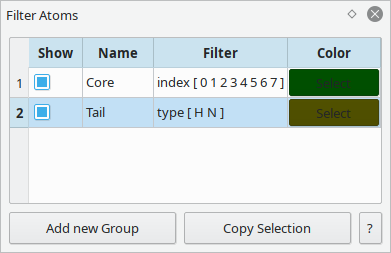
Select groups of atoms that meet the given criteria.
These groups can be used for visual highlighting or as a target for scripting.
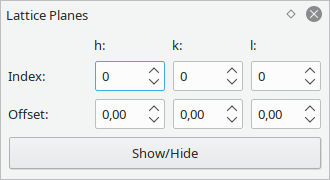
Display lattice planes according to their miller indices.
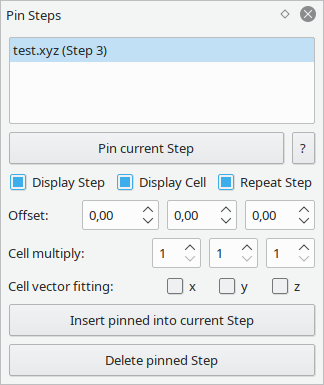
This tool allows to "pin" steps so they can be overlaid over other structures.
This can e.g. be used to track the time-evolution of a trajectory by pinning a specific configuration.
It is not limited to steps of a single trajectory though. You can e.g. import a molecule into a structure of a crystal surface to create surface adsorbates. With the multiplication and vector-fitting features, you can also easily create densely packed adsorption patterns.
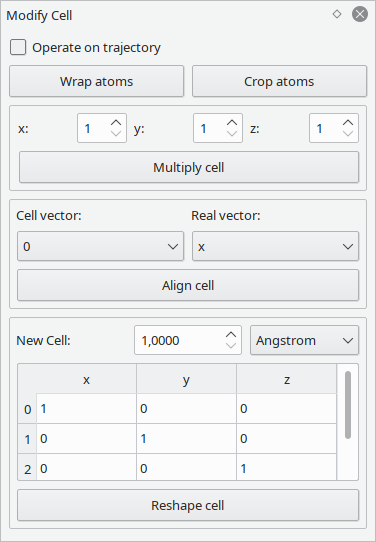
This tool provides a bunch of often-used operations on periodic structures.
These operations all consider the whole cell. For operations like multiplication and reshaping, keep in mind that atoms outside the box may lead to unexpected behavior. Consider performing a wrap or crop operation first, and don't forget to check for overlapping atoms.
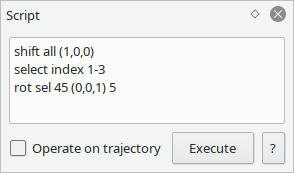
For precise modifications on groups of atoms, a simple scripting language is included.
Here you can shift, rotate and mirror atoms via absolute or relative coordinates.
This also interacts with the named groups: you can create groups in the script and use them as targets for operations.
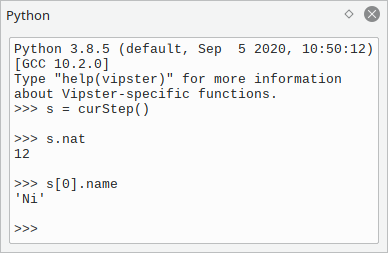
For more in-depth interactions, an embedded python widget is included.
This widget resembles a regular python shell with the Vipster module already loaded.
See the API documentation for details on general usage.
You can interact with structures loaded in the GUI via a few special functions
such as curMol() or curStep(),
please use the help() and dir() functions to find out more.
Note: this widget may be missing from your executable. Check the build instructions on how to build your own executable with the python widget.
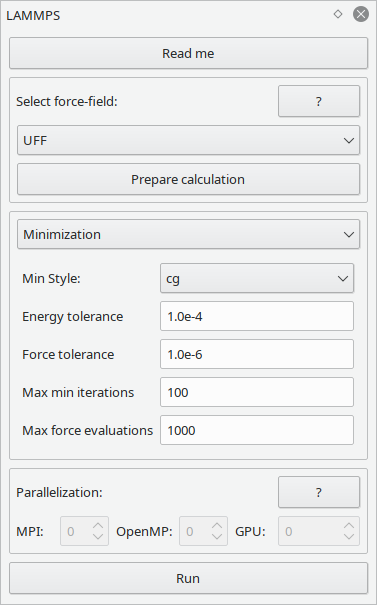
This widget allows to perform simple force field based simulations with the LAMMPS right inside Vipster.
Note: the integration is still in beta status and may lead to unexpected errors and crashes.
Performing simulations is a two-step process:
-
Set up the force field.
You can choose between embedded forcefields and a custom configuration. Embedded forcefields try to determine all atom and bond types as needed when you press the "Prepare calculation" button, and try to give you hints on how to modify your structure to fit the chosen forcefield. If this does not meet your needs, you can set your structure up by yourself (i.e. atom and bond types) and choose & configure the LAMMPS specific interaction styles.
-
Perform the calculation
If your structure is set up correctly, you can perform the simulation. You can choose between geometry optimizations and molecular dynamics, corresponding to LAMMPS'
minimizeandruncommands. Please see their documentation pages for further details.
Note: this widget may be missing from your executable. Check the build instructions on how to build your own executable with the python widget.